PCAC has three projects:
1.
Metabolic regulation of FOLFIRINOX acquired resistance in pancreatic cancer.
2.
MUC1 in Therapy Resistance.
3.
Targeting MARK2-HDAC signaling to overcome paclitaxel resistance in pancreatic cancer.

Research has shown that metabolic reprogramming is a targetable vulnerability to counteract acquired resistance in PDAC. The resistance involves both tumor cell-intrinsic mechanisms and metabolic and signaling crosstalk between tumor cells and the tumor microenvironment. Preliminary data from human PDAC patient-derived xenograft models identified peptidyl arginine deiminase 1 (PADI1) as a significantly upregulated gene associated with poor prognosis. PADI1 expression correlates with patient survival and is linked to the glycolytic phenotype and hypoxia gene signature in PDAC tumors. Inhibiting PADI1 activity has been shown to improve the responsiveness of PDAC cell lines and organoids to FOLFIRINOX therapy.
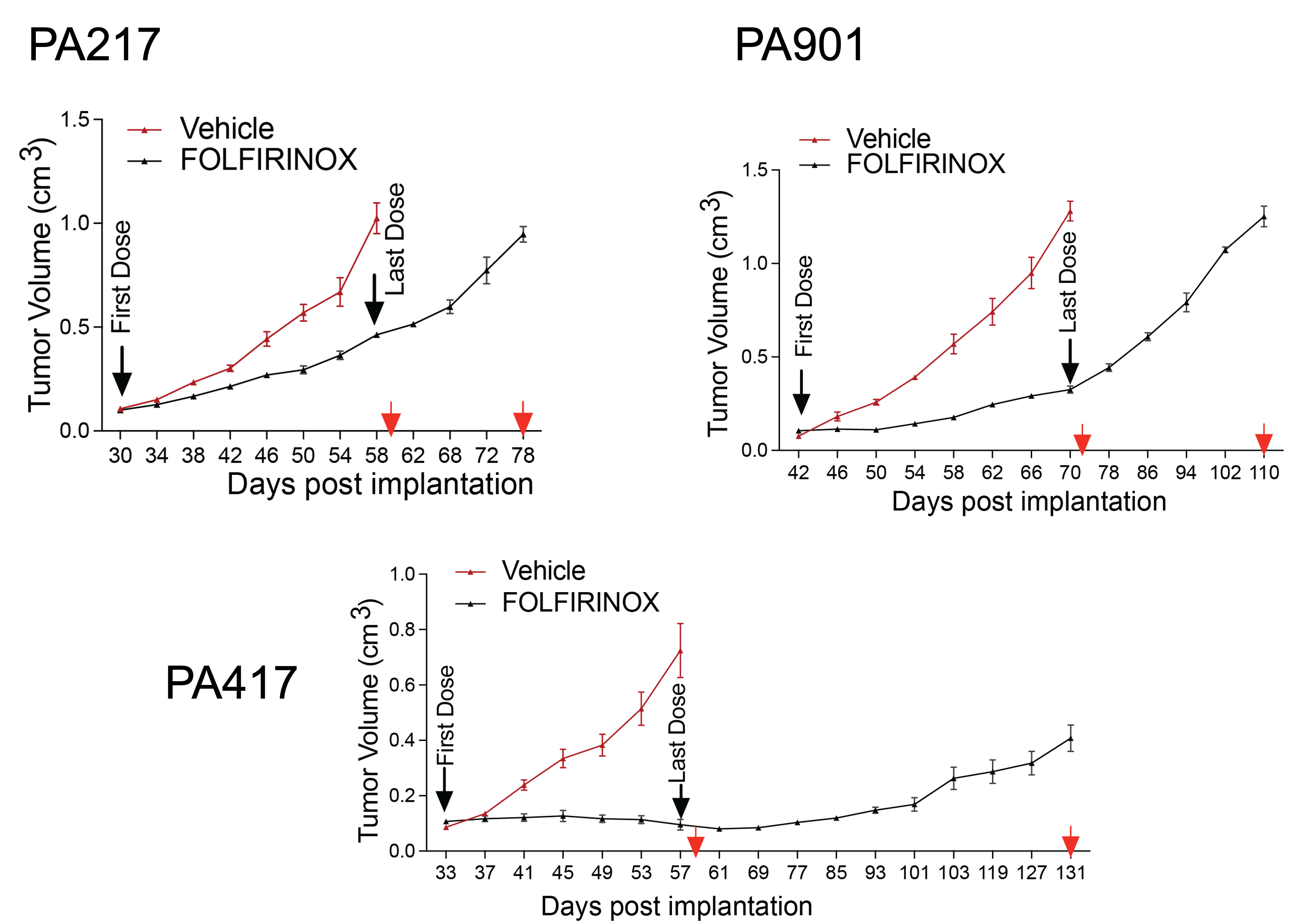
The proposed project will test the hypothesis that targeting PADI1 or its downstream metabolic pathways can prevent the development of resistance to FOLFIRINOX in PDAC. It will also explore the role of stromal remodeling in this resistance. The project is structured around three specific aims:
1.
Investigate the efficacy of targeting PADI1 downstream pathways and associated mechanisms of stromal remodeling for abrogating resistance to FOLFIRINOX therapy.
2.
Determine the mechanism of tumor-cell intrinsic metabolic reprogramming that also feeds into stromal reprogramming by PADI1.
3.
Investigate the efficacy of targeting the pathways identified in Aims 1 and 2 in PDX models and determine clinical correlates utilizing human tissue specimens.
These studies will provide new insights and strategies to target acquired resistance to FOLFIRINOX in PDAC.
(For additional information, please visit NIH RePORTER)

Pancreatic cancer is naturally resistant to therapy and quickly develops further resistance after treatment. High levels of the protein MUC1 have been shown to play a key role in both inherent and acquired resistance across multiple cancer types. MUC1 influences cancer cell behavior by stabilizing HIF-1a, which enhances glucose metabolism and contributes to drug resistance, particularly in pancreatic cancer treated with Gemcitabine.
Recent findings indicate that MUC1 is present on tumor-derived exosomes, which are selectively absorbed by various cells in the tumor environment, promoting tumor growth and drug resistance at both primary and metastatic sites. The overarching hypothesis is that exosomes from pancreatic cancer cells drive resistance to therapy by reprogramming tumor cells, cancer-associated fibroblasts, and immune cells. To explore this, the study proposes two specific aims:
Specific Aim 1.
Elucidate the molecular features of MUC1-positive exosomes that cause therapy resistance through reprogramming of tumor cells, cancer-associated fibroblasts, and immune cells at local or metastatic sites.
Specific Aim 2.
Evaluate expression signatures and pathways of therapy resistance in matched sets of primary tumors and metastatic lesions from untreated and treated patients (from our tissue core) by utilizing spatial transcriptomics (single-cell RNAseq) and by performing multiplexed immunofluorescence.
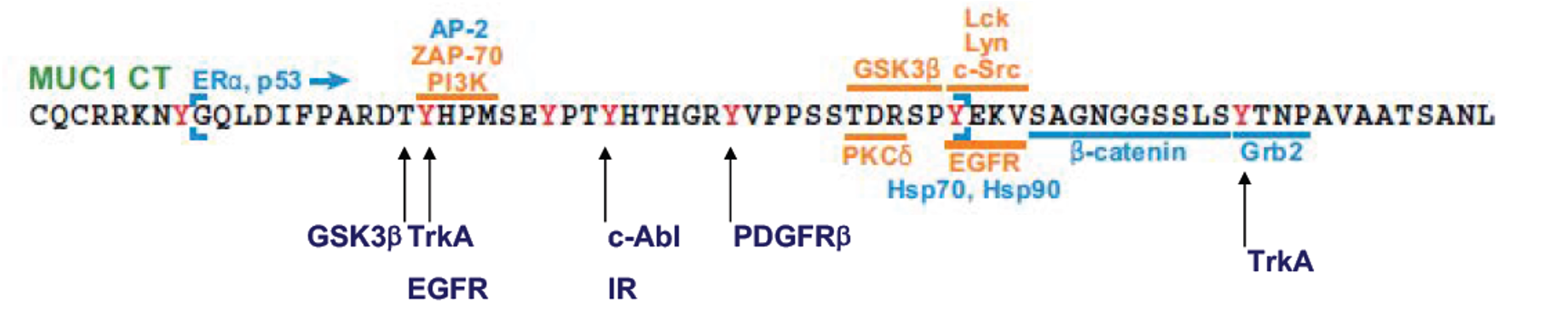
(For additional information, please visit NIH RePORTER)

Many antitubulin agents, such as paclitaxel (Taxol), are widely used to treat cancers like breast, ovarian, lung, and pancreatic. However, patient response varies greatly, and drug resistance is a significant challenge. Identifying prognostic markers to predict response and enhance drug sensitivity is crucial. Through kinome-wide screens, MARK2 (microtubule affinity-regulating kinase 2) was identified as a key regulator of Taxol sensitivity in pancreatic ductal adenocarcinoma (PDAC) cells. MARK2 is phosphorylated in response to antitubulin drugs, with the corresponding kinase and phosphorylation sites mapped. MARK2 influences Taxol's effectiveness without impacting normal cell growth and regulates chemosensitivity by controlling class IIa HDACs (histone deacetylases). Specifically, MARK2 phosphorylates HDAC4, which then regulates YAP (yes-associated protein) activity and the expression of Taxol-induced target genes. Inhibiting HDACs sensitizes PDAC cells to Taxol in both lab studies and animal models.

The central hypothesis is that the MARK2-HDACs axis could be a therapeutic target to overcome Taxol resistance in PDAC patients. This hypothesis will be tested through three specific aims:
Aim 1.
Determine the role and regulation of MARK2 in response to antitubulin chemotherapeutics;
Aim 2.
Elucidate the downstream effectors and mechanisms of MARK2 in response to Taxol chemotherapeutics;
Aim 3.
Targeting HDACs and serine metabolism to overcome Taxol resistance in PDAC. Identifying new regulators and signaling pathways triggered by antitubulin drugs could provide insights into chemoresistance mechanisms. The study suggests that combining HDAC inhibitors with antitubulin agents like Taxol may improve treatment efficacy in drug-resistant or recurrent PDAC patients.
(For additional information, please visit NIH RePORTER)

